朱致熹,张洁琳,陈依军
(中国药科大学生命科学与技术学院,南京 211198)
β-内酰胺类抗生素是临床使用最为广泛的抗生素[1]。由于抗生素的滥用,细菌耐药性已经严重影响β-内酰胺类抗生素的临床使用。特别是耐碳青霉烯类肠杆菌(carbapenem-resistant Enterobacte-riaceae,CRE),对用于临床治疗的大多数β-内酰胺类抗生素具有多重耐药性,其引发的感染性疾病治疗困难,并伴随高病死率[2]。依据美国疾病预防控制中心(CDC)数据报道,2017 年美国感染CRE的病例为13 100 例,造成1 100 例死亡,投入治疗经费超过1.3亿美金[3]。β-内酰胺类抗生素耐药性(特别是革兰氏阴性致病菌)已经成为威胁人类健康及造成社会经济损失的全球性挑战。
革兰氏阴性菌对β-内酰胺类抗生素产生耐药性主要原因是细菌β-内酰胺酶的表达。β-内酰胺酶可以水解该类抗生素的β-内酰胺环,造成抗生素效力下降甚至丧失,引发耐药性。β-内酰胺酶依据作用机制分为丝氨酸β-内酰胺酶(seineβ-lactamases,SBL)与金属β-内酰胺酶(metallo-β-lactamases,MBL)。SBL 依赖酶活性中心的丝氨酸残基,亲核进攻β-内酰胺类抗生素的β-内酰胺环使其失去活性[4]。MBL 依赖其活性中心的Zn2+,通过活化1分子水分子产生的氢氧根离子,对β-内酰胺环进行破坏[5]。MBL 分为B1、B2 和B3 3 个亚类。B1和B3亚类活性中心有两个Zn2+结合位点(Zn1和Zn2),B2 亚类只有1 个Zn2+结合位点[6]。临床相关的MBL 主要属于B1 亚类,如亚胺培南酶(imipenemase,IMP)[7]、维罗那亚胺培南酶(verona imipenemase,VIM)[8]和新德里金属β-内酰胺酶(New Delhi metallo-β-lactamase,NDM)[9]。B1亚类MBL 广泛存在于革兰氏阴性条件致病菌中,包括大肠埃希菌、肺炎克雷伯菌、铜绿假单胞菌与鲍曼不动杆菌等。
包括NDM-1在内的MBL在多重耐药菌中的表达,已成为威胁人类健康的重要问题。MBL 具有广泛的底物谱,可水解包括青霉素类、头孢菌素类以及被誉为抗生素“最后一道防线”的碳青霉烯类在内的几乎所有β-内酰胺类抗生素[10]。编码MBL的基因具有高传播性,可通过质粒以及其他可移动遗传元件(如转座子)在菌株间快速传播,使原本不具有抗性的菌株获得抗性[11]。此外,MBL 易变异,活性更强、稳定性更高的新突变体不断出现。临床急需针对表达MBL的多重耐药菌的有效治疗策略。
β-内酰胺类抗生素与β-内酰胺酶抑制剂的复方制剂,是治疗耐药菌感染的有效方法。β-内酰胺酶抑制剂抑制细菌β-内酰胺酶活性,可使细菌重新对β-内酰胺类抗生素敏感[12]。代表性药物有阿莫西林/克拉维酸、哌拉西林/他唑巴坦和头孢他啶/阿维巴坦等,但它们均用于治疗表达SBL 耐药菌引起的感染,对MBL 引发的耐药性感染无效。相较于SBL 抑制剂的开发与临床应用,MBL 抑制剂的开发起步较晚。自1990 年起,文献中报道的MBL 抑制剂已有上千种,但目前没有成功批准上市的化合物。
依据对MBL 抑制机制的不同,目前报道MBL抑制剂主要包括变构抑制剂、共价抑制剂、金属配体类抑制剂、螯合剂类抑制剂等[13]。其中螯合剂类MBL 抑制剂是最早进行开发的一类MBL 抑制剂[14],已有多个结构类型的化合物,可对临床常见的MBL 具有低至微摩尔浓度甚至纳摩尔浓度级别的半数抑制浓度(IC50)或抑制常数(Ki)。部分化合物在体外与体内实验中可以有效恢复碳青霉烯类抗生素对产MBL 耐药菌的抗菌效力,展现出与抗生素协同抗菌作用。螯合剂类MBL抑制剂对MBL抑制机制主要有两种:(1)通过螯合作用剥离或隔绝MBL 活性中心Zn2+,使MBL 失活或降解[15];
(2)进入MBL 活性中心,形成稳定的MBL∶Zn(II)∶抑制剂三元复合体,抑制MBL活性[16]。
螯合剂作为MBL抑制剂参与临床MBL耐药菌治疗的选择性与使用安全性一直受到关注。经典螯合剂乙二胺四乙酸(EDTA)虽然为强效的金属螯合剂,但其对金属离子的螯合不具备选择性,进入体内会使大量生理金属依赖的酶类(如Ca2+、Mg2+依赖的酶)失活,引发严重不良反应[17]。近年来有多篇文章报道了一系列包括aspergillomarasmine A(AMA)和吡啶-2,6-二羧酸(DPA)在内的新型螯合剂类MBL 抑制剂。这些化合物除具备对MBL的抑制活性,还具备对MBL的选择性,安全性好。部分化合物表现出良好的药物开发潜力,其构效关系与机制研究为同类药物设计与开发提供了重要信息。
本文就螯合剂类MBL 抑制剂的研究进展进行综述,讨论其化学结构、抑制活性、与抗生素的协同作用、选择性和作用机制(本文讨论的化合物结构以及活性相关信息见表1),为后续开发高选择性、高活性和低毒性的MBL抑制剂提供参考。

表1 螯合剂类MBL抑制剂的化学结构与活性相关信息
EDTA(1,图1)是使用最为广泛的合成类强效金属螯合剂。EDTA 具备螯合Zn2+能力,但因对其他生理金属酶的选择性差、毒性强,不能作为MBL抑制剂临床应用。EDTA的钙复合物(Ca-EDTA)是唯一获得临床批准的EDTA 类似物,主要用于治疗铅中毒。研究显示Ca-EDTA 可恢复亚胺培南对产IMP-1、IMP-2、IMP-7、IMP-10 及VIM-2 的铜绿假单胞菌的抗菌活性,对IMP-1 的IC50为55 μmol/L[18]。此外Ca-EDTA 还可提高亚胺培南和美罗培南对表达NDM-1 的肺炎克雷伯菌的抗菌活性(MIC 下降至小于2和小于4 μg/mL)[19]。
乙二胺-N,N′-二丁二酸(ethylenediamine-N,N′-disuccinic acid,EDDS,2,图1)是一种细菌来源的天然金属螯合剂,具有与EDTA 相似的螯合性质,可生物降解[20]。Proschak 等[21]报道了EDDS 对B1 亚类MBL 具有抑制作用,其对NDM-1 抑制活性最强(IC50为0.18 μmol/L),对VIM-1 和SIM-1 较弱(IC50均为8 μmol/L),对IMP-1 无效。EDDS 与亚胺培南协同,可恢复多种携带MBL的临床分离革兰氏阴性菌菌株对亚胺培南的敏感性。近期Tehrani等[22]报道酶法合成多种EDDS衍生物,体外实验显示6 种衍生物具有NDM-1 抑制活性(IC50均为微摩尔级),其中一种甲基取代衍生物表现出较高的美罗培南协同抗菌作用。

图1 EDTA及其相关化合物、AMA及其衍生物化学结构(A);
AMA螯合Zn2+示意图(B)[27]
AMA(3,图1)及相关化合物是近几年受到关注的新型螯合剂类MBL 抑制剂。作为一种真菌天然产物,1965 年AMA 首次从米黄曲霉菌中分离与鉴定,其具有使植物叶片枯萎、斑变等毒素活性[23]。在20 世纪80 年代,AMA 被报道为血管紧张素转化酶(ACE)抑制剂[24]。2014 年,King等[25]在Nature上报道,AMA 被重新鉴定为一种选择性金属β-内酰胺酶抑制剂,其对NDM-1 和VIM-2 具有低至微摩级的抑制活性(对NDM-1 的IC50为4 μmol/L,对VIM-2 的IC50为9.6 μmol/L)。通过对229 种临床分离的MBL 阳性致病菌的抑菌实验,发现AMA 可有效恢复美罗培南对携带NDM-1 和VIM-2 的多种革兰氏阴性菌(肠杆菌、不动杆菌、假单胞菌)的抗菌活性。体内实验显示,在注射致死剂量的NDM-1阳性肺炎克雷伯菌的小鼠模型中,单次联合注射美罗培南(10 mg/kg)与AMA(30 mg/kg)5 d 存活率达95%,而单独给药美罗培南或AMA均对致死性感染无效。
在后续的一项研究中,King 同一课题组继续考察AMA 与6 种β-内酰胺类抗生素联用对19 种B1、B2 和B3 亚类MBL的抑制活性[26]。相较于其他β-内酰胺类抗生素,碳青霉烯类抗生素尤其是美培罗南、多尼培南和亚胺培南与AMA的协同抗菌活性最佳。AMA 对B1 亚类的NDM-1 和VIM-2表现出最强抑制活性,对NDM-4、IMP-1、IMP-7 等B1 亚类MBL 具有中等抑制活性,对B2 亚类CphA和B3 亚类AIM-1 的抑制活性低,对所有SBL 没有抑制活性。值得注意的是,AMA活性与MBL的Zn2位点的Zn2+亲和力表现出相关性,对亲和力低的MBL抑制活性更高。
对AMA 作用机制的研究表明,AMA 通过选择性螯合Zn2+,导致MBL 活性中心低亲和力Zn2位点Zn2+的丢失,抑制MBL 活性(图1-B)。电感耦合质谱实验发现AMA 从1 分子NDM-1 中仅移除1 个Zn2+。Bergstrom 等[27]利用平衡透析和电感耦合等离子体原子发射光谱法(ICP-AES),发现AMA可以在体外有效移除NDM-1、VIM-2 或IMP-7 活性中心的Zn2+,在微摩尔浓度条件下,2当量的AMA可以移除1 当量的Zn2+。近期研究发现,包括NDM-1 在内的许多MBL 中Zn2位点较Zn1位点对Zn2+亲和力更弱,因此研究者推测AMA可能主要通过移除Zn2位点的Zn2+发挥抑制作用。此外,顺磁核磁分析显示,NDM-1 活性中心并没有形成AMA∶Zn(II)∶NDM-1三元复合体,表明AMA 可能并不直接进入活性中心。2021 年的最新研究进一步支持了这一推测,AMA不结合于NDM-1的活性中心,而是通过螯合游离的Zn2+,增强了动态发生的Zn2位点Zn2+的解离,导致Zn2+丢失并诱发蛋白质降解,从而导致NDM-1失活[15]。AMA 具有金属离子选择性,对Zn2+、Ni2+和Co2+具有螯合作用,而对其他生理重要的Mg2+、Ca2+等作用微弱,这可能是AMA 具有较低动物毒性(小鼠静脉注射给药的LD50为159.8 mg/kg,EDTA的LD50为28.5 mg/kg)的原因之一[15,25]。
自2016 年起,一系列文章报道了AMA 及结构类似物的合成方法。AMA 结构较为复杂,具有3 个手性中心及多个游离的氨基和羧基基团,化学全合成较为困难。至今共有6种主要的AMA 合成策略,包括4种全化学合成、1种化学酶偶联合成及1 种全生物合成方法[28-33]。在早期合成研究中,Liao 等[28]和Koteva 等[29]全化学合成4 种AMA的立体异构体,验证了真菌合成的天然AMA 为(S,S,S)-AMA。2018 年,本课题组成员参与研究并报道AMA 及其衍生物的化学酶偶联合成方法,通过引入一种高立体、区域与化学选择性的生物催化剂EDDS 裂解酶,缩短合成路线,获得了AMA 及一系列相关化合物[32]。2021 年,Guo 等[33]首次从米曲霉菌中鉴定并表征了AMA合成酶。该酶催化以L-天冬氨酸和O-磷酸丝氨酸(或O-乙酰丝氨酸)为底物连续两步反应合成AMA,为AMA 及其衍生物提供了一种酶催化合成方法。
在对约30 种AMA 衍生物的MBL 抑制活性考察中,部分衍生物(toxin A,4;
AMB,5)(图1)具有NDM-1 的抑制活性,抑制效力达AMA 同一数量级[22,34]。值得注意的是,AMA 结构中的羧基对其发挥抑制作用非常重要,在对羧基进行修饰后观察到其对NDM-1 活性丧失[31,34]。AMA的3 个手性中心的绝对构型对NDM-1 的抑制活性影响较小,(S,S,S)-AMA的IC50在所有衍 生物中最低[34]。AMA 衍生物的合成与活性考察促进该类化合物对MBL 抑制机制的研究,将为开发新的选择性MBL抑制剂提供重要参考。
1,4,7 -三氮杂环壬烷-1,4,7-三乙酸(NOTA,6,图2-A)与1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸(DOTA,7,图2-A)是胺类金属螯合剂。2015 年,Somboro 等[35]首次报道NOTA 和DOTA 可协同美罗培南或亚胺培南,恢复其对表达NDM、VIM 或IMP的耐碳青霉烯类抗生素的耐药菌的活性。其中,NOTA 效果略优于DOTA,4 μg/mL 质量浓度下的NOTA 可使美罗培南对多重耐药菌的MIC 降至0.06 μg/mL。细胞毒性实验显示,NOTA和DOTA在使用剂量下不会诱发溶血。
Zhang 等[36]报道一种NOTA的二硫代氨基甲酸酯衍生物(化合物8,图2-A)为MBL 抑制剂。体外实验显示,化合物8可以恢复美罗培南对多种携带blaNDM-1基因的临床分离菌株的活性,但活性低于NOTA。对NOTA 及化合物8的抑制机制考察显示,向携带NDM-1 的大肠埃希菌同时加入美罗培南与化合物8 可引起细菌生长率大幅下降(由100%下降至2.55%),体系中补加ZnCl2后耐药菌生长率由2.55%恢复至90.30%。相应的NOTA实验中也表现出相同现象,显示出NOTA 与化合物8对NDM-1 的抑制活性可以通过补加Zn2+得到逆转,作用机制可能与基于螯合作用的Zn2+剥离有关。动力学研究显示NOTA 和化合物8 对NDM-1的抑制符合非竞争性抑制的特征。分子对接结果显示NOTA 和化合物8 均可能不结合于NDM-1 的活性中心,因此作者推测该类化合物可能通过螯合从活性中心动态解离的Zn2+发挥抑制NDM-1活性。
Li 等[37]利用NOTA 对NDM-1 抑制活性,开发了一种复合纳米材料IPM@AgNPs-PEG-NOTA(图2-B)。该纳米材料将SH-PEG2000-NOTA 包裹于载有亚胺培南的银纳米颗粒外层,利用NOTA 逆转细菌对亚胺培南的耐药性,发挥银纳米颗粒与亚胺培南的双重抑菌作用。该纳米复合材料在体内与体外实验中表现出有效杀灭产NDM-1的耐碳青霉烯类抗生素的鲍曼不动杆菌的活性,相较于单独使用亚胺培南或银钠米颗粒的抗菌活性均得到增强。IPM@AgNPs-PEG-NOTA具有良好的生物相容性,毒性低,为多功能抗耐药菌药物的开发提供了新思路。
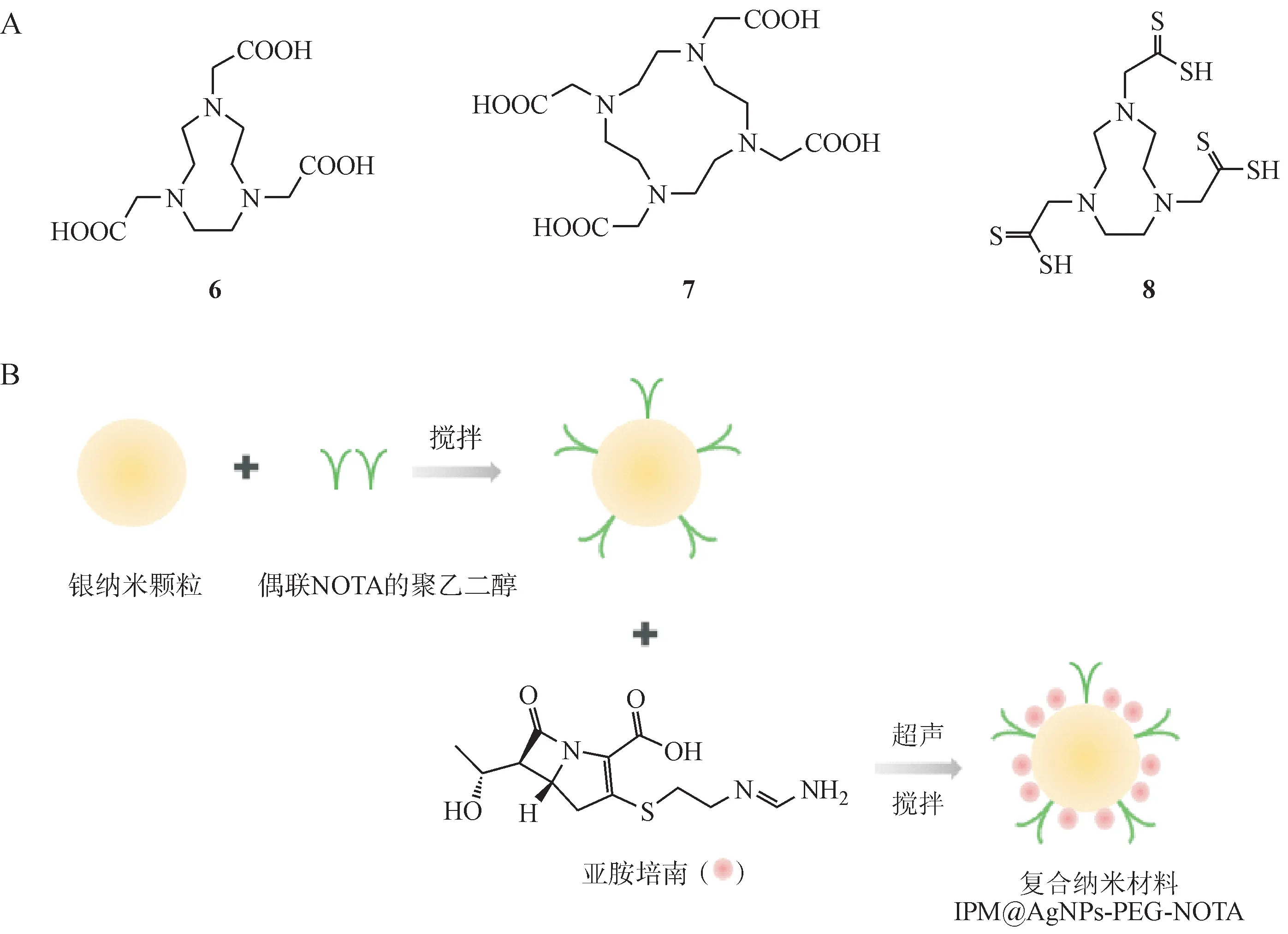
图2 NOTA相关MBL抑制剂化学结构(A);
IPM@AgNPs-PEG-NOTA复合纳米材料制备示意图(B)[37]
2-吡啶甲酸(2-PDCA,9,图3)是一种人体产生的天然Zn2+螯合剂,可帮助锌的吸收[38]。2007 年,Horsfall 等[39]首次报道2-PDCA 和吡啶-2,4-二羧酸(2,4-PDCA,10,图3)是B2 亚类CphA的竞争性抑制剂。X 射线与晶体分析结果显示,2,4-PDCA 通过在活性中心形成CphA∶Zn(II)∶2,4-PDCA 复合体的方式螯合Zn2+,使酶失活。值得一提的是,2-PDCA 对B1 亚类NDM-1、VIM-2、IMP-1 和B3 亚类L1没有抑制活性(IC50>100 μmol/L)。
2017 年,Chen 等[16]报道了吡啶-2,6-二羧酸(DPA,11,图3)是一种具有NDM-1 及其他相关MBL(VIM 和IMP)抑制潜力的化学结构。该课题组通过使用基于片段的药物发现(fragment-based drug discovery,FBDD)策略,设计、合成并评估了3组共47 个DPA 衍生物,发现衍生物(12)是一种有潜力的MBL抑制剂。衍生物12对NDM-1的IC50低至80 nmol/L(DPA的IC50为0.41 μmol/L),并对B1亚类的VIM-2 和IMP-1 同样有较强的抑制活性(对VIM-2 的IC50为0.21 μmol/L,对IMP的IC50为0.24 μmol/L)。体外实验中,衍生物12(100 mg/L)可使亚胺培南对临床分离的携带blaNDM-1大肠埃希菌和肺炎克雷伯菌的MIC 由4 ~ 16 mg/L 降低至0.5 ~1 mg/L,与亚胺培南具有协同抗菌作用。衍生物12(10 μmol/L 以下)对Zn2+依赖金属酶HDAC-1、HDAC-6 和hCAII 没有明显抑制活性,表现出对B1亚类MBL的高选择性,并且对人源HEK293 细胞没有细胞毒性[16]。机制研究结果显示,DPA 类似于EDTA,通过无选择性的螯合NDM-1活性中心的两个Zn2+发挥抑制作用,但衍生物12 则通过结合于NDM-1 活性中心,形成稳定的NDM-1∶Zn(II)∶衍生物12三元复合物发挥抑制作用。随后一项研究表明,衍生物12 在NDM-1 活性中心的结合主要源于苯胺的氨基与NDM-1 的211 位赖氨酸残基的相互作用[40]。
Hinchliffe等[41]报道将DPA的一个羧基置换为磷酰甲基基团,合成了一系列DPA 衍生物并考察对MBL 抑制活性。化合物13(图3)及另外两种衍生物对NDM-1、VIM-2、IMP-1 和L1 具有竞争性抑制活性(IC50为0.3 ~ 7.2 μmol/L,Ki为0.03 ~ 1.5 μmol/L),在对真核细胞不产生毒性的浓度下,可有效恢复美罗培南对临床分离的携带VIM-2、NDM-1 的肺炎克雷伯菌、铜绿假单胞菌、大肠埃希菌等耐药菌的活性。共晶结构显示化合物13 与IMP-1及L1的结合方式不同(图4-A 和图4-B)。在IMP-1 活性中心中,化合物13 的吡啶N 原子、羧基与Zn2位点Zn2+形成配位键,而磷酸基团与桥联的水分子相互作用,形成稳定的三元复合体。但在L1 的活性中心中,化合物13 的磷酸基团置换Zn2位点的Zn2+及氢氧根离子,形成单Zn2+形式酶以发挥对L1的抑制作用。
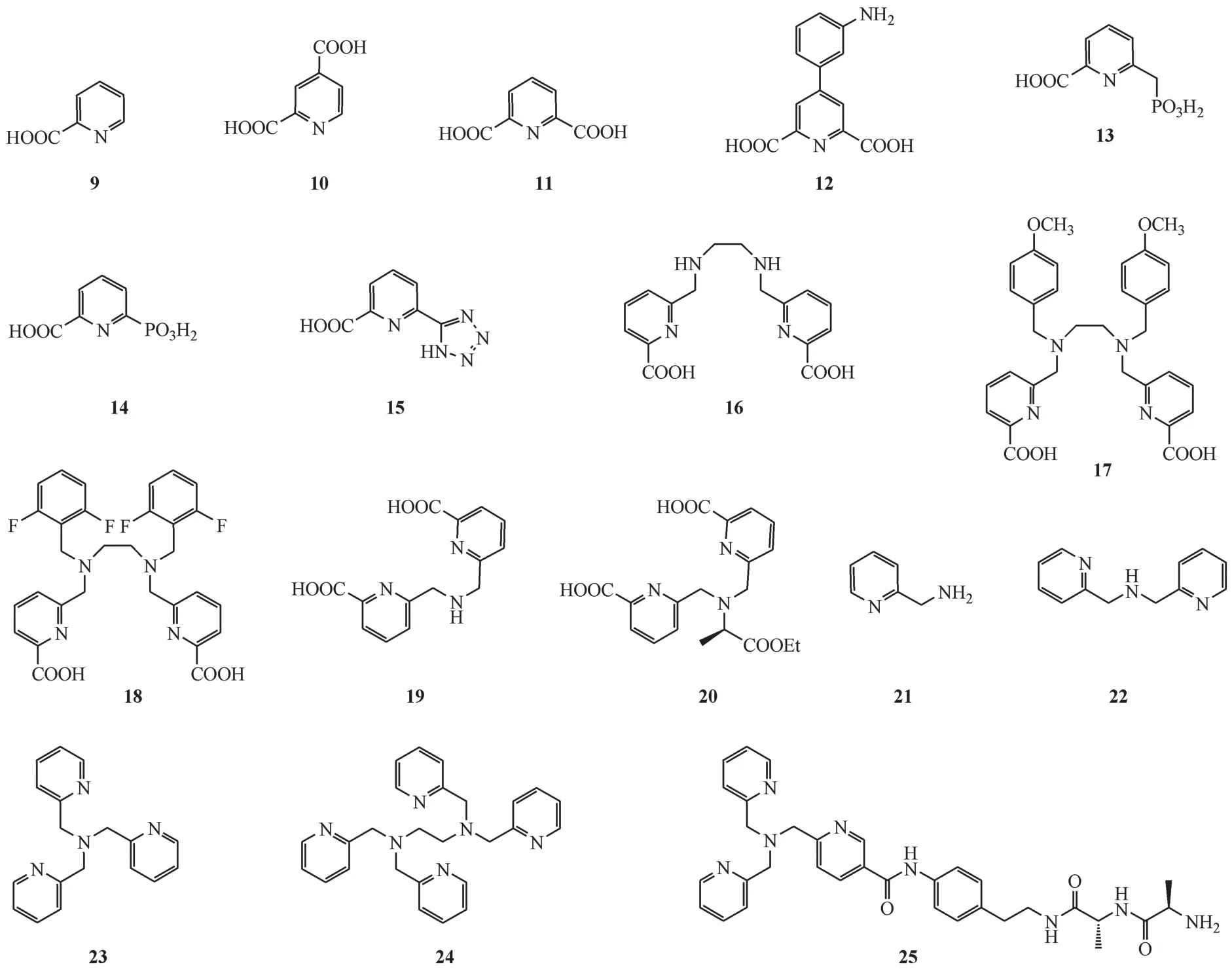
图3 吡啶羧酸类与吡啶甲基胺类MBL抑制剂
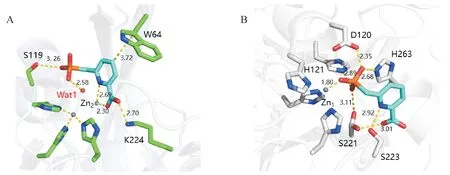
图4 化合物13与IMP-1共晶结构(PDB号:5HH4)(A)[41];
化合物13与L1共晶结构(PDB号:5HH5)(B)[41]
依据生物电子等排体置换思路,Chen 等[42]将DPA 其中一个羧基置换为其他电子等排体,合成一系列DPA 衍生物。结合抑制活性与平衡透析实验结果发现,当DPA 中羧基替代为其他电子等排体时,会导致化合物对NDM-1抑制机制发生改变。羧基替换为磷酸基团(14)时,其抑制机制仍为Zn2+的剥离,对NDM-1 的IC50下降至0.31 μmol/L。但当羧基替换为四氮唑(15)时,抑制机制改变为三元复合体的形成,对NDM-1 的IC50提高至7 μmol/L。该研究显示电子等排体置换策略是改变化合物对MBL 作用机制的有效方案,可为后续MBL 抑制剂的开发提供参考。
6,6′-[亚乙基双(亚胺亚甲基)]双(2-吡啶甲酸)(H2dedpa,16,图3)同属于吡啶羧酸的衍生物,可用作68Ga正电子发射断层显像剂的无环螯合剂,用于疾病诊疗[43]。Shi 等[44-45]鉴定H2dedpa 为强效的MBL 抑制剂(对NDM-1 的IC50为0.17 μmol/L),并以该结构为母体合成一系列衍生物,并从中找寻到活性更强的化合物17(对NDM-1 的IC50为0.04 μmol/L)和 化 合 物18(对NDM-1 的IC50为0.11 μmol/L)。此类化合物可在体外恢复美罗培南对临床表达NDM、IMP 菌株的活性,对NDM-1的IC50均小于1 μmol/L。为研究此类化合物抑制MBL机制,作者进行了Zn2+敏感性实验。在实验组耐药菌培养中同时添加美罗培南与化合物18,观察到细菌生长率大幅下降(由100% 下降至2.69%)。对实验组额外加入与化合物18 等摩尔量的ZnCl2,耐药菌生长率回复到正常生长的对照组相似水平。结果显示此类化合物对NDM-1的抑制活性受到Zn2+存在而发生逆转,以此判断此类化合物通过螯合Zn2+发挥抑制作用。分子对接结果显示此类化合物可结合于NDM-1活性中心。
Chen 等[46]报道6-{[(6-羧基吡啶-2-基)甲基氨基]甲基}吡啶-2-羧酸(H2dpa,19)衍生物为强效MBL 抑制剂,并通过Zn2+螯合机制对MBL 产生抑制作用。其中化合物20 对B1 亚类的NDM-1 的Ki为1.59 μmol/L,对IMP-1 的Ki为1.57 μmol/L,对VIM-2 的Ki为6.16 μmol/L。体内外实验均显示20可恢复美罗培南对携带NDM-1肺炎克雷伯菌的活性,且在有效浓度内未显示出溶血活性与对胃上皮细胞(GES)的细胞毒性。
吡啶甲基胺类化合物2-吡啶甲胺(MPA,21,图3)、二-(2-吡啶甲基)胺(22,图3)、三-(2-吡啶甲基)胺(TPA,23,图3)和N,N,N′,N′-四-(2-吡啶甲基)乙二胺(TPEN,24,图3)被鉴定为MBL 抑制剂,可在体外恢复携带NDM、VIM的耐碳青霉烯类抗生素肠杆菌对美罗培南敏感性[47]。此类化合物抑制机制为Zn2+螯合,并发现此类化合物结构中吡啶基团数目越多,其螯合Zn2+能力越强,对MBL 抑制活性越强[48]。
TPA 在后续研究中,用作螯合剂组件参与合成一系列全新的选择性Zn2+螯合剂。该螯合剂设计基于“Chelator+Linker+Modulator”思路,将TPA与一短肽片段偶联,调节亲脂性以降低毒性[49]。研究人员测定各化合物与美罗培南的协同抗菌活性,并考察各化合物对HepG2细胞的细胞毒性。在50 μmol/L浓度下,衍生物25 与TPA 均使美罗培南对携带NDM-1 肺炎克雷伯菌MIC 降至0.125 μg/mL。衍生物25 的细胞毒性更低,其对HepG2 细胞的IC50>100 μmol/L(TPA的IC50为9.8 μmol/L),同时在128 mg/kg 剂量下对小鼠单周给药两次未见不良反应。该设计策略保留TPA 对MBL 抑制活性,并降低了毒性。
除上述几类化合物,还有一些难以归类的螯合剂类金属MBL 抑制剂,如二硫代氨基甲酸酯类化合物[50]、吡唑类化合物[51]、支化聚乙烯亚胺(BPEI)及其衍生物[52]、喹啉类化合物[53]、螺二氢吲哚-噻二唑类似物[54]等。
Ishii 等[55]报道ME1071(马来酸衍生物,26,图5)可以体外增强碳青霉烯类抗生素对产MBL铜绿假单胞菌的活性。体外实验测得ME1071对IMP-1的Ki0.4 μmol/L,对VIM-2 的Ki120 μmol/L。体内实验结果显示,在注射携带MBL的铜绿假单胞菌引起的肺炎模型小鼠中,ME1071 可协同比阿培南有效治疗肺炎,且未产生不良反应[56]。
Gavara 等[57-59]报道通过修饰1,2,4-三唑-3-硫酮的4 位及5 位位点,合成了一系列衍生物。此类化合物在体外明显增强美罗培南对产VIM的临床分离肺炎克雷伯菌的活性。化合物27(图5)是VIM-2的竞争性抑制剂,Ki为0.83 μmol/L。化合物27 与VIM-2 的共晶结构(图5-B)显示,该化合物可同时螯合VIM-2 活性中心两个Zn2+。其中化合物27 的2 位N 原子螯合VIM-2 的Zn1位点Zn2+,硫酮螯合Zn2位点Zn2+,4 位和5 位的修饰与活性中心其他氨基酸发生相互作用稳定该三元复合体结构。
基于芳基磺酰腙化合物与头孢菌素的相似性,Shaaban 等[60]将其与吡唑类化合物偶联,理性设计NDM-1 的抑制剂,以期获得能稳定结合于NDM-1的活性口袋、螯合Zn2+以实现抑制作用的化合物。获得的衍生物在体外实验中均显示出优秀的对NDM-1、IMP-1、AIM-1 的抑制活性。衍生物28(图5)活性最强,可使头孢氨苄和美罗培南的活性提高约99.8%。体内实验中,对感染肺炎克雷伯菌的模型小鼠进行给药实验,相比于只给药美罗培兰的小鼠在5 d 内全部死亡,治疗组(美罗培兰+ 衍生物28)小鼠全部存活,并且6 d 内检测到体内所有细菌清除。衍生物28 在350 mg/kg 给药剂量下对小鼠无不良反应。分子对接结果与预期一致,衍生物28 结合于NDM-1 和IMP-1 活性中心,通过磺基螯合Zn2+发挥抑制活性。
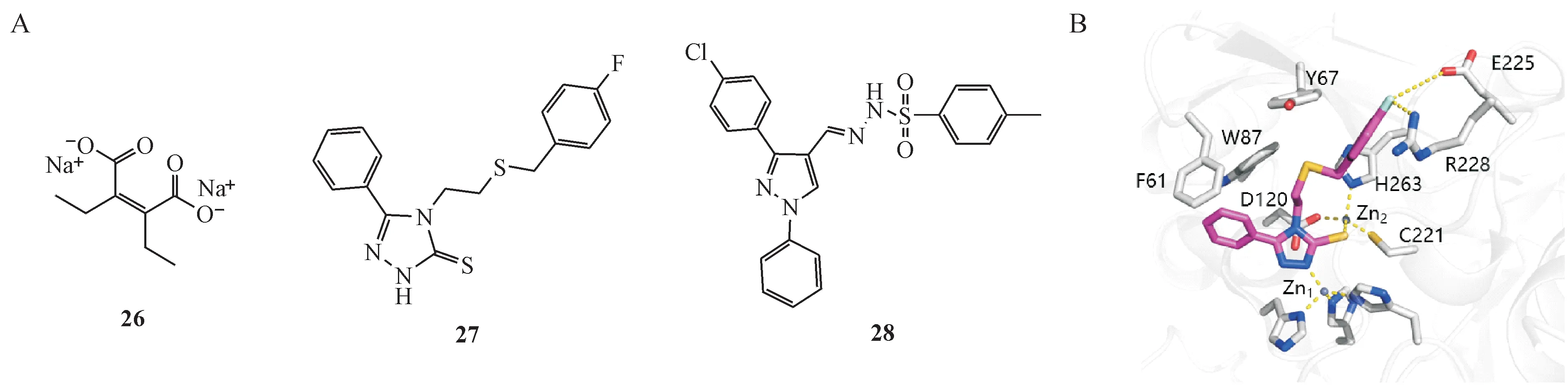
图5 其他螯合剂类MBL抑制剂(A);
化合物27与VIM-2共晶结构(PDB号:7OVF)(B)[57]
本文综述了螯合剂类MBL 抑制剂的研究进展,介绍了几类重要化合物的化学结构、抑制活性、与抗生素的协同作用、选择性和作用机制。
螯合剂类MBL 抑制剂主要对B1 亚类的MBL(NDM、VIM、IMP)表现出有效的体外抑制活性(IC50或Ki达微摩尔级)。其中吡啶羧酸类化合物的IC50或Ki更低,部分化合物达到纳摩尔级别。抑制剂可在体外协同碳青霉烯类抗生素,恢复携带MBL的耐药菌对碳青霉烯类抗生素的敏感性。部分化合物在体内活性实验中展现出与碳青霉烯类抗生素的协同作用,但大部分化合物缺乏对体内活性的考察。
氨基多羧酸类抑制剂的作用机制主要为Zn2+剥离。相比于广谱螯合剂EDTA,近期得到关注的AMA表现出更高的动物耐受性(小鼠静脉注射给药的LD50为159.8 mg/kg)。原因可能为AMA 是一种选择性金属螯合剂,对Zn2+螯合能力强于其他生理金属离子如Mg2+、Ca2+、Mn2+等。由于NDM-1和VIM-2 等MBL 对Zn2+亲和力比许多生理Zn2+依赖酶弱,AMA 对Zn2+的纳摩尔级亲和力(Kd为0.2 nmol/L)能够移除这些MBL的Zn2+,减少对高亲和力Zn2+依赖酶的脱靶效应。AMA的作用机制为同类抑制剂的开发及结构改造提供了一定参考。吡啶羧酸类及吡啶甲基胺类抑制剂的作用机制包括Zn2+剥离与形成稳定MBL∶Zn(II)∶抑制剂三元复合体。对DPA 衍生物的研究显示结构修饰可以改变作用机制,并且同一抑制剂可能对不同MBL 具备不同的抑制机制(如化合物13)。代表性衍生物12经改造由Zn2+剥离机制变为形成三元复合体机制,抑制活性提高,同时在有效浓度下对几种生理金属酶无抑制活性,显示了靶向活性中心的设计改造可能是一种有效策略。目前许多报道集中于发现高活性抑制剂,缺乏抑制剂对生理金属酶选择性、作用机制、构效关系、体内活性与毒性等研究。后续对这些方面的系统考察,将为螯合剂类MBL 抑制剂的开发提供重要参考。
对现有螯合剂进行理性设计,提高螯合剂对Zn2+的选择性,降低毒性可能是此类药物未来的发展方向。已有研究人员进行相关尝试,通过对化合物22 偶联靶向MBL的细菌二肽D-Ala-D-Ala,降低细胞毒性,提高对MBL的抑制活性[61]。未来研究中也可能通过制剂手段提高螯合剂类MBL 抑制剂的靶向性与成药性,前文报道的NOTA的复合纳米材料不仅具有良好生物相容性,还提高了NOTA的抑菌活性。随着更多螯合剂类MBL 抑制剂的鉴定与开发、各类化合物MBL 抑制机制的不断解析、理性设计与制剂方法的不断尝试,相信不久后高选择性、高活性和低毒性的螯合剂类MBL 抑制剂将与β-内酰胺抗生素联用,解决临床表达MBL的耐药菌感染问题。

(续表)
猜你喜欢 吡啶衍生物抑制剂 PD-1抑制剂联合仑伐替尼治疗晚期肝癌的疗效分析中国典型病例大全(2022年11期)2022-05-13血管紧张素转换酶抑制剂在肾内科的应用分析健康体检与管理(2022年4期)2022-05-13质子泵抑制剂对反流性咽喉炎的疗效研究医学概论(2022年4期)2022-04-244个进口PD-1/PD-L1抑制剂不良反应信号的挖掘与评价中国药房(2022年7期)2022-04-14有机硼敲开吡啶药物合成“新大门”科学导报(2022年21期)2022-04-102,3-二氟-5-氯吡啶和3,5-二氯-5-氟吡啶高效液相色谱分析现代农药(2022年1期)2022-02-152—氯吡啶的合成及应用成长·读写月刊(2017年3期)2017-04-08烃的含氧衍生物知识测试题中学生数理化·高二版(2016年3期)2016-12-26烃的含氧衍生物知识链接中学生数理化·高二版(2016年3期)2016-12-26咪唑吡啶类化合物的合成方法进展科技视界(2016年26期)2016-12-17